Una caracterización genómica exhaustiva de líneas derivadas de tumores malignos de los nervios periféricos desafía los criterios diagnósticos actuales
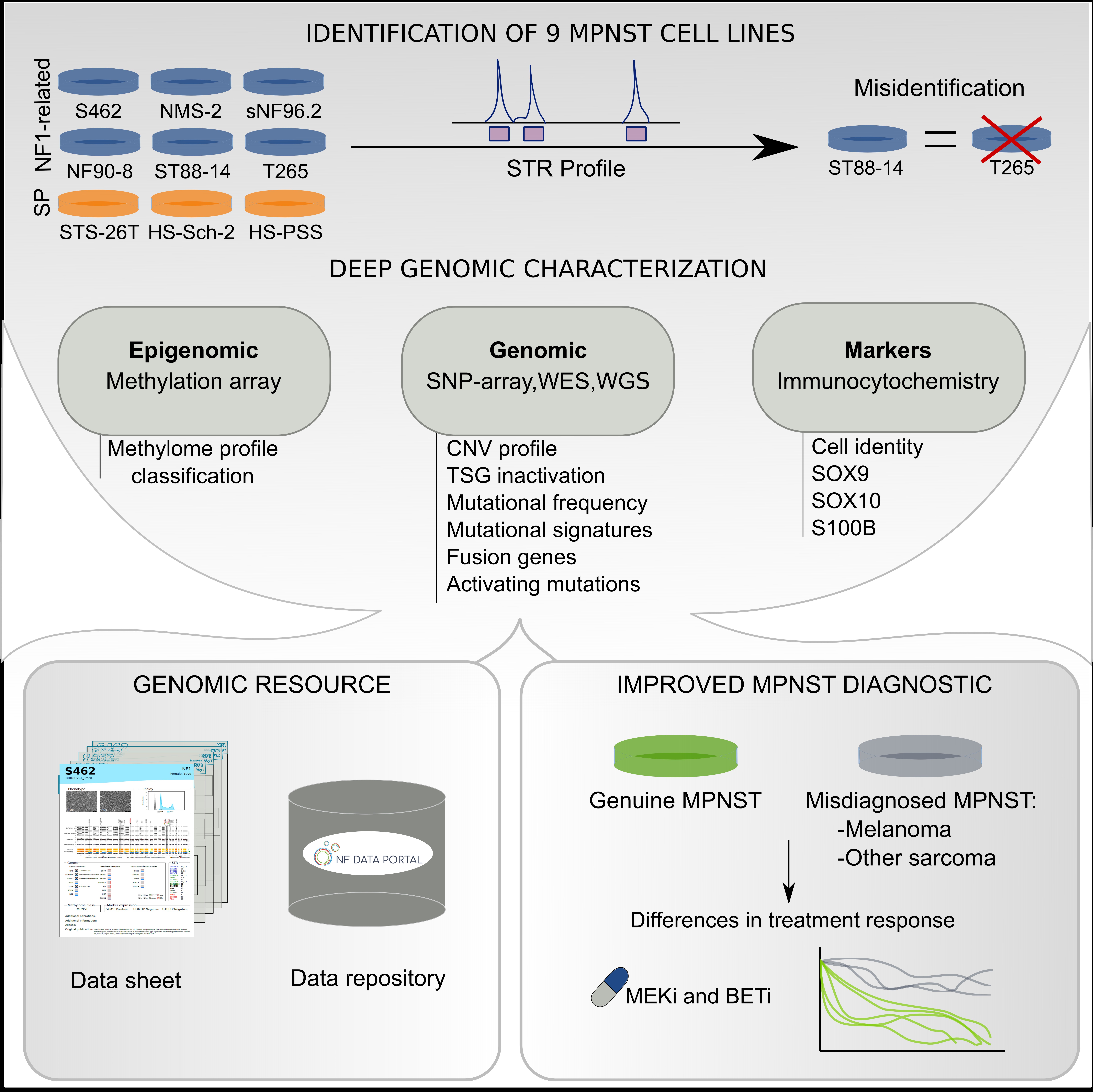
El grupo de investigación de Cáncer Hereditario del IGTP ha liderado una colaboración internacional que ha permitido la caracterización genómica de las líneas celulares más comúnmente utilizadas derivadas de tumores malignos de las vainas de los nervios periféricos (MPNSTs). Esta investigación ha permitido generar un catálogo detallado de alteraciones genómicas para cada línea celular, que podrán utilizarse para desarrollar estrategias en terapias de precisión. A la vez, se ha creado un repositorio genómico abierto a todos los investigadores interesados en estos tumores. El trabajo ha sido publicado en la revista iScience.
Uno de los resultados más destacados es que aporta nuevos tipos de información genómica que complementa la caracterización histológica de los MPNSTS en aras de un mejor y más preciso diagnostico diferencial de estos tumores. De hecho, el trabajo ha permitido identificar que algunas de las líneas supuestamente MPNSTs, que se han utilizado en múltiples trabajos de investigación, o que están depositadas en bancos de líneas celulares (ATCC, RIKEN), no son en realidad MPNSTs, si no otras entidades tumorales.
La Dra. Miriam Magallón, primera firmante del trabajo, comenta "hemos identificado líneas celulares que estaban mal diagnosticadas, pero también hemos detectado que dos de las líneas más utilizadas como MPNSTs, son en realidad, la misma línea celular".
Los Dres. Bernat Gel y Eduard Serra, los investigadores que han liderado este trabajo, resaltan que "este trabajo es una gran oportunidad para utilizar los nuevos tipos de información genómica aportada para revisar los criterios diagnósticos de estos tipos tumorales. Nos gustaría liderar un nuevo proyecto internacional donde patólogos, biólogos moleculares y bioinformáticos, pudiéramos analizar y comparar de manera sistemática la histología de estos tumores, conjuntamente con los datos genómicos de los mismos, para esclarecer qué es un MPNST, si existen diferentes subtipos, y qué no lo es".
Bernat Gel destaca finalmente "este estudio también abre la puerta al desarrollo de nuevos análisis genómicos que faciliten el manejo de los pacientes que tienen un alto riesgo de desarrollar un MPNST, o que ya lo han desarrollado, y requieren de un seguimiento periódico. Los análisis basados en biopsia mínima o biopsia líquida, con la utilización de nuevas tecnologías de secuenciación de lecturas largas serían ideales para identificar el tipo de alteraciones genómicas responsables del desarrollo y progresión de los MPNSTs".
El Grupo de Cáncer Hereditario del Instituto de Investigación Germans Trias i Pujol (IGTP) hace años que estudia tumores del sistema nervioso periférico asociados a la Neurofibromatosis de tipo 1, especialmente la progresión que va de neurofibroma plexiforme (PNF), a neurofibroma atípico (o ANNUBP, en inglés), hasta tumor maligno de la vaina de los nervios periféricos (MPNST, en inglés). Los dos pilares de la caracterización de estos tumores por parte de este grupo, son el análisis genómico, epigenómico y bioinformático, por un lado, y por el otro el desarrollo de modelos celulares que representen fidedignamente estos tumores. En relación a este punto, el Grupo de Cáncer Hereditario del IGTP también acaba de publicar en la revista STAR Protocols un trabajo donde detalla un protocolo experimental para la generación de neurofibromaesferas, un modelo 3D derivado de células madre de pluripotencia inducida (Mazuelas et al. 2023. DOI: 10.1016/j.xpro.2023.102198).
En este estudio han colaborado los laboratorios responsables de establecer estas líneas celulares, investigadores de 12 laboratorios distintos, de 5 países diferentes (Alemania, Bélgica, Japón, Estados Unidos y España). El trabajo ha sido financiado por el Instituto de Salud Carlos III (PI17/00524, PI20/00228), la Fundación Proyecto Neurofibromatosis y la Fundació La Marató de TV3 (51/C/2019).
Ayuda de $600,000 para hacer un ensayo pre-clínico con una nueva estrategia terapéutica para tratar los neurofibromas cutáneos
El grupo de investigación ha recibido una ayuda de $615,000 de la Neurofibromatosis Therapeutic Acceleration programo (NTAP)-Johns Hopkins University School of Medicine para llevar a cabo un proyecto destinado a probar una nueva estrategia terapéutica por los neurofibromas cutáneos en pacientes con Neurofibromatosis de Tipo 1. El proyecto está liderado por el grupo de Cáncer Hereditario, pero se realiza conjuntamente con el Grupo del Dr. Piotr Topilko, Mondor Institute for Biomedical Research (Francia).
Las personas con NF1 pueden desarrollar de decenas a miles de neurofibromas cutáneos a su piel. Estos tumores benignos que se originan en las terminaciones nerviosas que hay entre la epidermis y la dermis, no representan un peligro por la vida de estas personas, pero en cambio, tienen un impacto en su calidad de vida muy alto. Estos tumores pueden ser desfigurantes, molestos, y pueden cubrir el cuerpo de una persona, afectando de manera muy significativa su autoestima, la socialización, etc.
El grupo de Cáncer Hereditario descubrió que la combinación de agentes que elevan el AMPc intracelular, conjuntamente con inhibidores de MEK (vía de Ras/MAPK), tiene un gran impacto en la viabilidad tanto de células de Schwann primarias derivadas de estos tumores, como en un modelo 3D de neurofibromaesferas, derivado de células madre de pluripotencia inducida (iPSCs). Con estos prometedores resultados a nivel in vitro se presentó un proyecto dentro de la iniciativa de NTAP "Biology and Therapeutic Development (BTD) for Cutaneous Neurofibromas (cNFs) Application" para hacer el salto a dos modelos animales complementarios y recoger información pre-clínica in vivo respecto a este co-tratamiento. Uno de estos modelos ha sido desarrollado por el Dr. Piotr Topilko a Paris y el otro es un modelo PDX que resulta de injertar en el nervio ciático de ratones las neurofibromesferas derivadas de iPSCs, en colaboración con el Grupo de la Dra. Conxi Lázaro y la Dra. Juana Fernández, del IDIBELL.
El proyecto durará tres años y estará liderado por la Dra. Helena Mazuelas, tanto en Barcelona como París, y coordinado por la Dra. Meritxell Carrió y el Dr. Eduard Serra.
Referencia
Miriam Magallón-Lorenz et al. Deep genomic analysis of malignant peripheral nerve sheath tumor cell lines challenges current malignant peripheral nerve sheath tumor diagnosis. iScience. 2023 Jan 31;26(2):106096. DOI: 10.1016/j.isci.2023.106096. PMID: 36818284; PMCID: PMC9929861.